- ホーム
- >
- 細胞生物学用語集トップ
- >
- 新着細胞生物学用語集(%E3%83%86%E3%82%AF%E3%83%8E%E3%83%AD%E3%82%B8%E3%83%BC)
新着細胞生物学用語集(%E3%83%86%E3%82%AF%E3%83%8E%E3%83%AD%E3%82%B8%E3%83%BC)
| s-FDAP法 |
|---|
| 【s-FDAP (sequential FDAP) analysis】 |
| 木内 泰 |
| 東北大学大学院生命科学研究科 分子生命科学専攻 単分子動態生物学分野 |
| お問合せ |
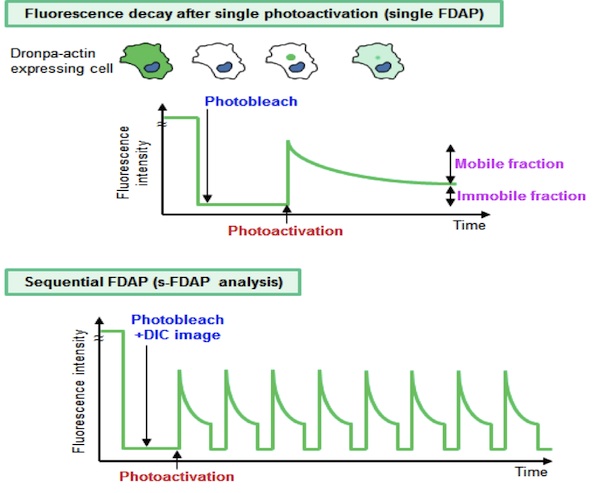
Dronpaは、AndoらによってクローニングされたGFP類似の単量体蛍光タンパク質である(1)。蛍光特性として、488nmの強いレーザー光で蛍光が消え(消光)、405nm又は458nmのレーザー光でその蛍光が回復し(光活性化)、光によって蛍光のスイッチのオン・オフを可逆的に繰り返すことができる。Dronpaをタグに付けたアクチンを細胞に発現させ、細胞全体で消光した後に一部で光活性化し、その後の蛍光強度の減衰を測定すれば、その減衰量は細胞質でのG-アクチン量を反映している(single FDAP)。そしてこの消光、光活性化、蛍光減衰の測定及び微分干渉像取得を繰り返すことで、細胞質のG-アクチン濃度の経時変化と細胞の形態変化を同一の細胞でモニターすることができる(s-FDAP analysis)(2)。
参考文献(1) Science, 306: 1370-1373, 2004. (2) J. Cell Biol., 193: 365-380, 2011. |
| 腸管ループアッセイ |
|---|
| 【ligated intestinal loop assay】 |
| 大野 博司 |
| 理化学研究所・横浜研究所 免疫アレルギー科学総合研究センター(RCAI) |
| お問合せ |
麻酔下の実験動物を用いて、生体組織の応答を、直接的に観察する実験手法。主に、パイエル板を含む小腸で行われる。まず、小腸のパイエル板を含む部分の両端を結紮し、生体条件に近い状態の閉空間を作る。この閉空間に試薬や菌液を充填し、組織との作用を観察する。組織が生きた状態でしか見られない応答(エンドサイトーシス等(1))を観察するのに適した手法である。参考文献(1)Hase, et al. nature (2009) |
| 培養基質の制御と細胞 |
|---|
| 【Regulation of culture substrate and cellular function】 |
| 原田 伊知郎 |
| 東京工業大学大学院生命理工学研究科 |
| お問合せ |
| 臓器・組織から取り出した細胞は培養液中で浮遊もしくはプラスチックシャーレの底に接着させて培養するのが一般的である。接着培養する細胞は、シャーレの底に物理吸着した細胞外マトリクス(以下ECM: Extracellular matrix) にインテグリン(Integrin)という膜タンパク質を介して接着している。したがって、細胞とECMとの接着の分子機構自体は生理的なものである。しかし、細胞外マトリクスが吸着しているプラスチックシャーレが生理条件下ではあり得ないほどの固いため、その環境は生体内とは著しく異なっていると考えられている。このような接着している細胞の足場(scaffold)の固さが細胞の機能に直接影響を及ぼしているのではないかという配慮から、近年様々な柔らかい培養基質が考案されている。 培養基質の固さの制御方法として細胞生物学的基礎研究によく用いられているのは電気泳動に用いるアクリルアミドゲルを培養基質とする方法である。アクリルアミドゲルは作成する際に添加する架橋剤の濃度を調整するだけで、容易に様々な固さのものを作成することができる。しかし、このゲルはタンパク質がほとんど吸着しないため、細胞外マトリクスもほとんど吸着しない。そのため、方法は様々であるがアクリルアミドゲルとECMとを架橋剤を介して結合させるのが一般的である。この培養基質を用いた研究によって、通常シャーレに培養した細胞に見られる極端な細胞骨格の発達や、アメーバのように広がった細胞の形はシャーレの固さが原因であったことが示されつつある。また、培養基質の固さを調整することで、細胞の分化能や増殖能などの機能も制御できることが示されている。 他にも様々な培養基質が考案されているものの、入手しやすいアクリルアミドゲルが多く用いられているもう一つの理由として、ゲルの透明度が上げられる。そのため細胞が観察しやすく広く普及している。特に、このゲル中に直径0.2〜1μmのマイクロビーズを包埋しておくと、ビーズの動きからゲルの歪みを計測することが出来るため、細胞が足場に力をかけている様子までも観察できる。そのため、細胞は移動に伴いどのような力を足場にかけているのか、等のような基礎研究にも本培養基質が用いられており、現在はTraction Force Microscopyとよばれ、新しい解析手法として普及している。 細胞が足場に加える力の制御・計測する他の方法として、ECMのマイクロパターニングやマイクロピラーを用いた研究も報告されている。カバーガラスにECMをプリントし、その形に細胞の形態を制御することで細胞骨格の発達を抑制し、柔らかい足場と同等の効果をねらった研究が報告されている。また、アクリルアミドゲルを用いたTraction Force Microscopyでは、接着点に直接かかっている力の定量化は逆問題であることから厳密な解析は難しい。そこで、3次元的なマイクロパターニング技術により、マイクロピラーをシリコンゴム(PDMS: polydimethylsiloxane)で作成して培養基板とすることで、一つ一つの接着点にかかる力のイメージングも試みられている。 参考文献(1) Pelham, R.J., and Y.L. Wang. 1997. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 94:13661-13665.
(2) Munevar, S., Y. Wang , and M. Dembo. 2001. Traction force microscopy of migrating normal and H-ras transformed 3T3 fibroblasts. Biophysical Journal. 80:1744-1757. (3) Engler, A.J., S. Sen, H.L. Sweeney, and D.E. Discher. 2006. Matrix elasticity directs stem cell lineage specification. Cell. 126:677-689. (4)Tan, J.L., J. Tien, D.M. Pirone, D.S. Gray, K. Bhadriraju, and C.S. Chen. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci USA. 100:1484-1489. |
| 原子間力顕微鏡 |
|---|
| 【Atomic Force Microscopy】 |
| 芳賀 永 |
| 北海道大学大学院生命科学院 |
| お問合せ |
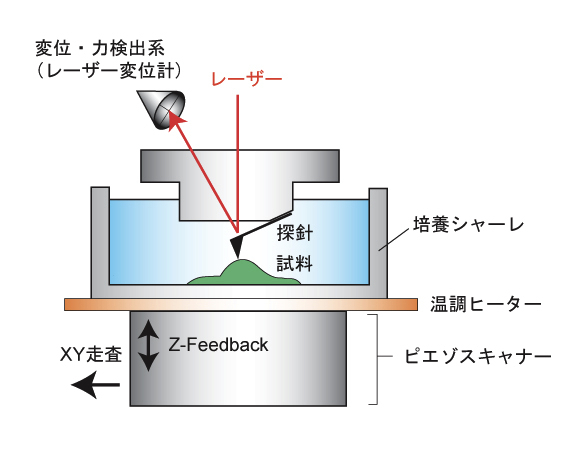
| 原子間力顕微鏡(Atomic Force Microscopy; 以下 AFM)は、生体試料の表面形状を測定することができる装置である。AFMは電子顕微鏡とは異なり、試料の固定処理を必要とせず、タンパク質や細胞の3次元的な形状を培養環境下で直接観察することが可能である。 また、カンチレバーとよばれる板バネの先端に長さ数ミクロン程度の針がついた探針を試料表面に直接接触させながら形状測定を行うため、カンチレバーのなぞり方を工夫することで、試料の形状だけではなく力学的性質(弾性率や粘着力など)の計測が可能となる(図)。 現在、AFMを用いた弾性率測定の主流はコンタクトフォースモードと呼ばれる測定法である。コンタクトフォースモードとは、探針を試料に押し込むことでカンチレバーに発生する力と試料の変形量との関係(フォースカーブ)を測定し、試料の弾性率を定量的に計測する測定法である。例えば、測定領域を64×64ピクセルに分割して、各点でフォースカーブを取得し、ピクセルごとに得られた弾性率を画像化(マッピング)することで硬さの空間分布像を得ることができる。 また、タンパク質分子のN末端とC末端をそれぞれ基盤とカンチレバーに結合させ、両端を引っ張ることにより発生する力を計測することで、タンパク質分子の折り畳みエネルギーや分子内のドメイン構造に関する情報を得ることができる。 この他、試料表面の走査を高速化することで、タンパク質一分子の移動や形の変化をビデオレートでイメージングする技術も開発されている。 参考文献 |
| 超解像顕微鏡法 |
|---|
| 【Super-resolution Microscopy】 |
| 岡田 康志 |
| 理化学研究所 神戸研究所 生命システム研究センター(QBiC) |
| お問合せ |
| 光が波としての性質(回折)を示すために、無限に小さな光源であっても、その光学顕微鏡像は波長の半分程度の大きさに滲んでしまう。可視光の場合、その大きさは200~300nmであり、これにより光学顕微鏡の分解能は規定される(回折限界 diffraction limit)。
近年、蛍光分子の特性を上手く活用することで回折限界を超える高い分解能を達成する技術が複数開発され、超解像顕微鏡法(super-resolution microscopy)と総称されている。現在よく知られている超解像顕微鏡法には、大きく分けて3つの種類がある。 1. 蛍光分子局在顕微鏡法(fluorescence localization microscopy) PALM/STORM/GSDIM (Photo Activation and Localization Microscopy, STochastic Optical Reconstruction Microscopy, Ground State Depletion and Individual Molecule return)と様々な名前が付けられているが、その基本的な原理は共通で、視野内の蛍光分子をランダムに個別に点滅させることで超解像を達成する。同時に点灯している蛍光分子の間隔が十分広ければ、各蛍光分子の位置の決定精度は画像のS/Nによって定まるので、回折限界以上の精度を達成することが出来る。これを多数回繰り返すことで視野内の蛍光分子すべての位置を決定し、その位置情報から画像を構築することで、超解像蛍光像が再構成される。そのため、分解能は、蛍光分子の位置決定精度で定まり、30 nm 以上の分解能が達成できる (fig1 画像1=通常蛍光像、画像2=PALM像 試料:微小管)。 この方法の発展型として、蛍光分子の明滅や消退による蛍光強度の揺らぎが蛍光分子を単位とする確率的な現象であることを利用して、蛍光強度の変動から超解像蛍光像を再構成する手法も開発されている(SOFI: Super-resolution Optical Fluctuation Imaging) 2. 構造化照明法(SIM: Structured Illumination Microscopy) SIMは、モアレ干渉を利用して特定方向の高分解能成分を取得する方法である。3方向×5位相の合計15枚の画像から再構成することで、x,y,zすべての方向で通常の蛍光顕微鏡の約2倍の分解能が達成できる。従って、例えばGFPなどの場合120 nm程度の分解能が期待できる。これは他の超解像手法顕微鏡法に比べると低い分解能であるが、蛍光色素の制約がないので、容易に多重標識の3次元超解像像を得られるのが特徴である。画像3(fig2)は、SIMによるミトコンドリア像。ミトコンドリアの内部構造を見ることが出来る。 3. 誘導放出制御法(STED: Stimulated Emission Depletion) STEDは、レーザー走査型コンフォーカル顕微鏡(CLSM: Confocal Laser Scanning Microscopy)の発展型である。誘導放出を利用してレーザーで蛍光が励起される点の大きさを回折限界以下に絞り込むことで超解像を達成する。従って、通常のCLSMと同様の操作で高速超解像イメージングが出来る。分解能は誘導放出制御光強度の平方根で決まり、およそ約50~80nmである (fig3 画像4:通常CLSM像、画像5:STED像 試料:EYFP標識F-actin)。ただし、使用できる色素には、励起光、蛍光、誘導放出光の3つがそれぞれ異なる波長の条件を満たす必要があるため、多重染色は困難である。 参考文献 |


| フェルスター共鳴エネルギー移動 あるいは 蛍光共鳴エネルギー移動 |
|---|
| 【FRET (Förster Resonance Energy Transfer あるいはFluorescence Resonance Energy Transfer) 】 |
| 松田 道行 |
| 京都大学大学院生命科学研究科 生体制御学分野 |
| お問合せ |
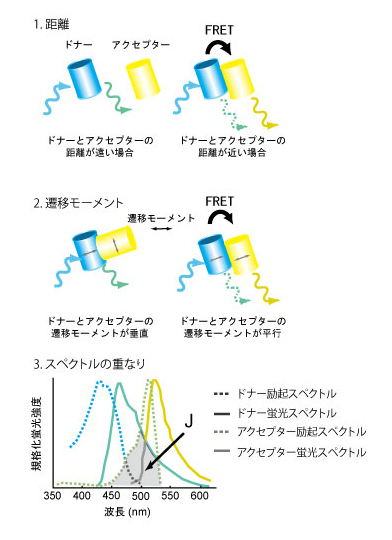
FRETとはこの現象の発見者の名前にちなんだFörster Resonance Energy Transferの略である。しかし、最近ではFluorescence Resonance Energy Transferの略と書いてある書籍が多く、蛍光共鳴エネルギー移動と邦訳されることが多い。FRETは、光子により励起されたドナー蛍光分子からごく近傍にあるアクセプター分子へエネルギーが共鳴移動する現象のことである。FRETの効率は、【1】蛍光発色団間の距離、【2】遷移モーメントの向き、【3】ドナー分子の蛍光波長域とアクセプター分子の吸光波長域の重なり、により主に規定される(1)。もっとも一般的なFRETの観察法はドナー蛍光の減少とアクセプター蛍光の増加を測定するが、厳密には、FRETはドナー分子の蛍光寿命を測定して定量される。FRETが蛍光タンパク質間でも観察されることを利用して、カルシウムを始めとするイオン、タンパク質リン酸化酵素、低分子量GTP結合タンパク質など、非常に多くの細胞内情報伝達分子に対するバイオセンサーが開発されている(2)。創薬においては、非常に長い蛍光寿命を持つユーロピウム(Eu)をドナーに用いることにより高いシグナルノイズ比のアッセー系が開発されている。参考文献 |
| 二光子励起顕微鏡法(多光子励起顕微鏡法) |
|---|
| 【Two-photon excitation microscopy, multi-photon excitation microscopy】 |
| 松田 道行 |
| 京都大学大学院生命科学研究科 生体制御学分野 |
| お問合せ |
二光子励起顕微鏡法とは、二光子吸収過程により分子を励起し、その蛍光を観察する顕微鏡法である。三光子励起も報告されているので多光子顕微鏡という用語を使う場合も多い。一分子が同時に二つの光子により励起される確率は自然界ではゼロといってよく、フェムト秒超短パルス高出力レーザが開発されて初めてこの現象が観察されるようになった。光密度が二光子励起を誘導するまでに高くできるのはレンズの焦点のみであるため、光路上の焦点以外に存在する分子は励起されない。これにより、二光子顕微鏡法では背景光がほぼなくなり、高いシグナルノイズ比が達成される。また、赤外レーザを使うため、組織透過性が高い。動作距離の長いレンズや、散乱を減少させる方法の開発により、脳組織では数mm程度の深部まで蛍光観察できるようになっている。近年、生きた生物で様々な細胞の動きや機能をリアルタイムに観察するintravital imagingという分野が発展しているが、その牽引力となっている技術である(1)。参考文献 |
| スピニングディスク共焦点顕微鏡 |
|---|
| 【spinning-disk confocal microscope】 |
| 中野 明彦 |
| 東京大学大学院理学系研究科生物科学専攻 |
| お問合せ |
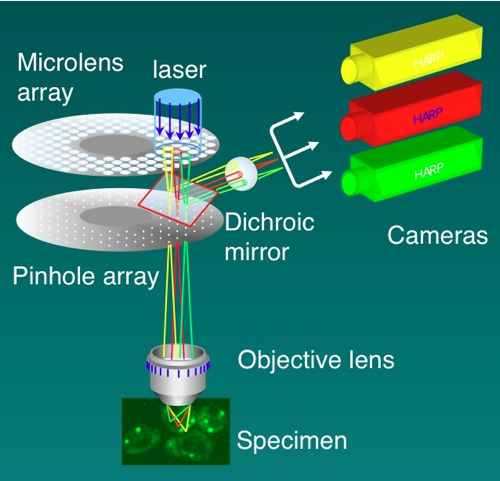
ニポウ板(Nipkow disk)式共焦点顕微鏡ともいう。小さな円盤に多数のピンホールを空け,高速で回転することによって試料を高速で走査する。横河電機が,ピンホールにレーザー光を効率よく導くためにマイクロレンズアレイを組み合わせて実用化した(図)。1000-2000フレーム/秒の高速性を誇る。同時に1000個程度の光点で試料を走査するため,ダメージが少ない,褪色しにくいなどの利点があり,また光学系は動かさないので実像を結び,高感度カメラで直接撮像できる。HARPカメラやEM-CCDカメラ,さらにイメージインテンシファイアなどとの組み合わせで,生細胞でも実際に1秒間に数百フレームの画像獲得が可能である。多色ダイクロイックミラー等を用いて複数種の蛍光プローブを完全同時観察でき,また高速性を生かしてオーバーサンプリングを行い,デコンボリューション処理することによって3Dで50-60 nmという超解像も実現されている。高性能なシステムは,SCLIM (Super-resolution Confocal Live Imaging Microscope)と呼ばれている。他の超解像顕微鏡に比べて,時間軸でも高い分解能を有することが特徴で,微細構造の高速な運動を追うライブセルイメージングの目的に適している。参考文献1. Nakano, A. (2002). Spinning-disk confocal microscopy -- a cutting-edge tool for imaging of membrane traffic. Cell Struct. Funct. 27:349-355. 2. Nakano, A. and Luini, A. (2010). Passage through the Golgi. Curr. Opin. Cell Biol. 22:471-478. 3. Matsuura-Tokita, K., Takeuchi, M., Ichihara, A., Mikuriya, K., and Nakano, A. (2006). Live imaging of yeast Golgi cisternal maturation. Nature 441:1007-1010. 4. Okamoto, M., Kurokawa, K., Matsuura-Tokita, K., Saito, C., Hirata, R., and Nakano, A. High-curvature domains of the endoplasmic reticulum (ER) are important for the organization of ER exit sites in Saccharomyces cerevisiae. J. Cell Sci. in press. |
| スペックル顕微鏡 |
|---|
| 【Fluorescent Speckle Microscopy】 |
| 渡邊 直樹 |
| 東北大学 大学院生命科学研究科 |
| お問合せ |
Waterman-StorerとSalmonがみいだした細胞骨格動態の蛍光顕微鏡撮影法。1996年、彼らが蛍光標識チュブリンを細胞にマイクロインジェクションしていたところ、低濃度で標識された細胞では、微小管がまだら(speckled)にみえ、微小管の軸方向への移動が可視化されたことの発見に端を発する。光の回折限界である0.2-0.3 ミクロンの領域に2-8個の蛍光標識が存在する場合、その数の確率的ばらつきから蛍光の強弱が生ずるが、それを可視化できる高感度の冷却CCDカメラが普及したことが本法の発見につながった。当初は、蛍光標識体のフォトブリーチング法よりも長時間にわたり、より高い時空間解像度で、微小管やアクチンネットワークの細胞内移動速度や方向を可視化する方法として利用された。後に、更に低密度の標識体をもちいて、細胞内アクチンを1分子ごとに可視化する単分子スペックル顕微鏡に発展し、アクチン線維の重合・脱重合やその制御分子の細胞内動態の可視化解析に用いられた。また、密度の高い標識の動態をコンピューター解析することで、細胞骨格分子や細胞接着分子の動態の全体像を迅速にとらえるqFSM法(quantitative Fluorescent Speckle Microscopy)がDanuserらによって開発された。(qFSM法については、結果に部分的にエラーが含まれる可能性も指摘されている。)参考文献 Salmon, E. D., Waterman, C. M. (2011) Mol. Biol. Cell. 22: 3940-3942.
Waterman-Storer, C. M., Salmon, E. D. (1997) J. Cell Biol. 139: 417-434. Watanabe, N., Mitchison, T. J. (2002) Science 295: 1083-1086. Danuser, G., Waterman-Storer. C. M. (2006) Annu. Rev. Biophys. Biomol. Struct. 35: 361-387. Vallotton, P., Small, J. V. (2009) J Cell Sci. 122: 1955-1998. |
| リポソームと細胞骨格(アクチン、微小管、セプチン) |
|---|
| 【Reconstruction of cytoskeletons (actin, microtubule or septin) using liposome】 |
| 滝口 金吾・滝口 陽子 |
| 名古屋大学大学院理学研究科 |
| お問合せ |
| 生体膜の形態形成や動態制御の機構をインビトロの系で研究する際、膜のモデルとして巨大人工脂質膜小胞(巨大リポソーム、giant liposome、giant vesicleまたはgiant unilamellar vesicleなど)がよく使われる。巨大リポソームは、脂質二重膜(lipid bilayer、脂質二分子膜など色々な呼び方がある)が水溶液中で自然に閉じてできる人工膜小胞(liposome またはvesicle)の中でも直径がμmオーダー以上のものをそのように呼ぶ [1, 2]。細胞と同程度の大きさであり、位相差、蛍光、微分干渉、暗視野などいろいろな光学顕微鏡法で直接観察ができる利点を持つ。 巨大リポソームの作製には、試験管内に作製したリン脂質を主成分とする脂質フィルムに水溶液を加え静置して得る静置水和法(gentle hydration、natural swellingなど)や [3-5]、白金や透明導電ガラス(ITO (indium tin oxide)ガラスが多く用いられる)の表面にリン脂質を主成分とする脂質を塗布しておき、水溶液中で交流電圧を印加して作るエレクトロフォーメーション法(electro formation)が主流である [6]。最近は、界面通過法(spontaneous transfer)など、油水界面にできる脂質の単層の膜(lipid monolayer、これも脂質一分子膜など色々な呼び方がある。単層の脂質二重膜との混同に注意!)を利用して作製する新たなリポソーム作製法も開発され、より広汎な条件下、より良い効率で巨大リポソームが得られるようになってきている [7, 8]。 細胞骨格と生体膜との相互作用を見る目的で、アクチンや微小管、セプチンなどを巨大リポソームに再構成させる場合には2通りある。 1つは、巨大リポソーム内に細胞骨格蛋白質を封入し、リポソーム内部の空間で再構成させる方法である。静置水和法やエレクトロフォーメーション法でリポソームを作製する際に、重合前のG-アクチンやチューブリンを含んだ水溶液を使うと、自然に内部にG-アクチンやチューブリンを取り込んだ巨大リポソームが形成されてくる。このリポソームの形成後に温度の上昇や塩の内部供給によって、リポソームの内部でアクチン線維や微小管の重合を起こさせることができる [3, 4, 9]。また界面通過法などの手法を用いれば、既に重合してできたアクチン線維やアクチンの束もリポソーム内部に再構成することが可能である [7, 8]。 もう1つは、予め作製しておいた巨大リポソームを含む溶液に、灌流装置を用いて細胞骨格蛋白質溶液を添加することにより、リポソームの外部から相互作用させ、その表面で再構成させる方法である。セプチンの膜結合能の検証および膜突起誘導活性の発見は、この方法によって成された [5]。 リポソームの作製は容易で観察法も様々あるので、膜の研究を行う際に大変有効である。しかし、巨大リポソームに限らず、リポソームを用いて実験しようとするときには以下のことに留意する必要がある。リポソームの作製にどのような手法を用いるにしても、脂質組成や作製時の温度、溶液条件が、作製効率ならびに作製されてくるリポソームのサイズや形態に大きく影響する。特に作製時に使う水溶液が2価の陽イオンや高濃度の塩を含む場合、多層の脂質二重膜からできた膜小胞(multilamellar vesicle)や、脂質二重膜を形成仕損なった脂質分子が凝集してできた油滴(lipid droplet)などが形成されてくるので、リポソームとの混同に注意しなければならない。生理的な塩濃度など高い塩濃度条件下でリポソームを使用したり作製したりするためには、先ず等張の糖を含む溶液で作製しておいたリポソームを実験に使う、脂質組成にPEG(polyethylene glycol)を結合させた脂質を加えて作製する、界面通過法などのような手法を用いてリポソームを作製する、などの工夫が必要である [5-9]。 参考文献[1] R. Lipowsky, Nature 349, 475-481 (1991)
[2] H. Hotani et al., Biosystems 71, 93-100 (2003) [3] M. Honda et al., J. Mol. Boil. 287, 293-300 (1999) [4] T. Kaneko et al., J. Mol. Boil. 284, 1671-1681 (1998) [5] Y. Tanaka-Takiguchi et al., Curr. Biol. 19, 140-145 (2009) [6] T. Wollert & J.H. Hurley, Nature 464, 864-869 (2010) [7] K. Takiguchi et al., Langmuir 27, 11528-11535 (2011) [8] K. Takiguchi et al., Methods Enzymol. 464, 31-53 (2009) [9] L. Limozin et al., Phys. Rev. Lett. 95, 178101 (2005) |
| 1分子イメージング法 |
|---|
| 【Single-molecule imaging technique】 |
| 原田 慶恵 |
| 京都大学 物質-細胞統合システム拠点(iCeMS) |
| お問合せ |
| 生体分子に様々な標識を付けて、それを手がかりに光学顕微鏡で個々の分子の局在や運動等を観察する手法を1分子イメージングあるいは1分子観察という。標識は大きく分けて2種類ある。大きな標識と、蛍光を発する標識である(蛍光1分子イメージング法の項を参照)。大きな標識を使う場合、1分子イメージングに特別な装置は不要で、光学顕微鏡で容易に観察できるという利点がある一方で、生体分子1個の観察ができているか、機能が損なわれていないかについての注意が必要である。大きな標識として直径1μm程度のマイクロビーズがよく使われる。マイクロビーズは光ピンセット(1分子操作法の項参照)で操作することや位置を精密に計測する実験(1分子操作法の項参照)にも使うことができ、1分子イメージングと1分子操作、1分子計測を組み合わせた実験を行うことができる。 |
| 蛍光1分子イメージング法 |
|---|
| 【Single fluorescent molecule imaging technique】 |
| 原田 慶恵 |
| 京都大学 物質-細胞統合システム拠点(iCeMS) |
| お問合せ |
| 蛍光色素、蛍光タンパク質、量子ドットなどの蛍光物質を個々の生体分子に標識して蛍光顕微鏡と高感度カメラを使って個々の分子を観察する方法である。特に、蛍光色素や蛍光タンパク質の場合は、微弱な蛍光を検出しなければならない。そのため、背景光を激減させることができるエバネッセント照明を組み込んだエバネッセント蛍光顕微鏡を使う。レーザーと光学部品を使ってエバネッセント照明の光学系を自作することもできるが、対物レンズ型エバネッセント照明を組み込んだ顕微鏡システムも市販されている。 エバネッセント照明法ではガラス基板から100nm程度の深さまでしか観察できないので細胞表面の観察は可能であるが、細胞の中を観察する実験には用いることができない。そこで考案されたのが斜光照明法である。斜光照明法を用いることで、細胞内の蛍光色素分子も1分子イメージングが可能になる。さまざまな蛍光プローブのうち、量子ドットは非常に明るく、退色しないので、in vitroの実験だけでなく、細胞内や個体内で1分子を追跡する実験に用いられている。 |
| 1分子操作法 |
|---|
| 【Single-molecule manipulation technique】 |
| 原田 慶恵 |
| 京都大学 物質-細胞統合システム拠点(iCeMS) |
| お問合せ |
| 生体分子の機能を調べるためには個々の分子を操作する手法が有効である。1分子を捕捉、操作する方法としてレーザー光を対物レンズで集光させ、直径1~数μm程度の微小粒子を光でトラップする「光ピンセット」、プラスチックの中に酸化鉄が封入された磁気ビーズや磁性粒子を磁石を使って操作する「磁気ピンセット」、原子間力顕微鏡(AFM)の力測定モードなどが使われる。光ピンセットや磁気ピンセットの最大捕捉力は100pN程度、AFMは固いカンチレバーを使うことで、数百pN以上の大きな力で捕捉することができる。 2つの光ピンセットを使って、2個のビーズを捕捉し、その間に1本のDNA分子を伸展させ、RNAポリメラーゼ分子の結合を観察する実験や、DNAの片端をガラス基板上固定し、もう一方の端には磁気ビーズに結合させ、磁気ピンセットでDNAをねじった後、磁気ビーズの高さ変化を計測することで、トポイソメラーゼがATPを加水分解し、ねじれを解消する反応を検出した実験など、光ピンセットや磁気ピンセットはDNAを使った実験に使われることが多い。また、AFMを使い、カンチレバーの先端にタンパク質分子を結合させ、それを引っ張ってタンパク質分子のドメイン構造が壊れていく様子などが観察されている |
| 1分子計測法 |
|---|
| 【Single-molecule measurement technique】 |
| 原田 慶恵 |
| 京都大学 物質-細胞統合システム拠点(iCeMS) |
| お問合せ |
| 生体分子に結合させた標識の位置や動きを高精度で1分子計測することで、個々の分子がどのようなメカニズムで機能しているのかを明らかにすることができる。
マイクロビーズなどの大きな標識は可視光で照明し、対物レンズでその像(影)を大きく(1000から10万倍)拡大し4分割のフォトダイオードに投影することで、その動きを1nm以下の精度で検出することができる。ただし、微小な動きを検出する場合は、標識のブラウン運動を防ぐために光ピンセット等で捕捉する必要がある。また、光ピンセットで捕捉することによって、そのバネ定数と検出した変位から、ビーズにかかっている力を見積もることができる。モータータンパク質であるキネシン分子を固定した直径1μmのビーズを光ピンセット(バネ定数~0.1pN/nm)で捕捉し微小管と相互作用させることで、キネシン分子が微小管に沿って8nmずつステップ状に動くことと最大7pNの力を出すことが明らかになった。また、RNAポリメラーゼがDNAの情報を1塩基ずつ読み進んでいく0.34nmステップの動きも同様の方法で検出された。 蛍光色素分子や量子ドットなどを1分子イメージング蛍光顕微鏡で観察した場合、直径が数百nmのぼやけた像として観察されるため、そのままでは、正確な位置はわからない。しかし、この像の光強度のプロファイルを解析することによってその中心点、すなわち蛍光物質の存在している位置をnm精度で決定することができる。この方法を使って二量体のモータータンパク質分子であるミオシンVが2つのモータードメインを交互に動かすことによってアクチンフィラメントに沿って運動することが明らかになった。 最近は量子ドットを使って、細胞内で1分子計測が行われ始めている。 |





